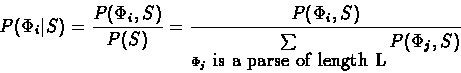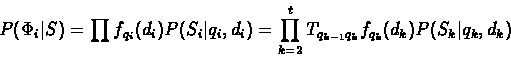As we have seen, a Hidden Markov Model (HMM)
is a Markov chain in which the states are not directly observable.
Instead, the output of the current state is observable. The output
symbol for each state is randomly chosen from a finite output
alphabet according to some probability distribution.
A Generalized Hidden Markov Model (GHMM) generalizes the HMM as
follows: in a GHMM, the output of a state may not be a
single symbol. Instead, the output may be a string of finite
length. For a particular current state, the length of the output
string as well as the output string it self might be randomly
chosen according to some probability distribution. The probability
distribution need not be the same for all states. For example,
one state might use a weight matrix model for generating the
output string, while another might use a HMM. Formally a GHMM is
described by a set of four parameters:
A finite set Q of states.
Initial state probability distribution .
Transition probabilities Ti,j for .
Length distributions f of the states (fq is the length distribution for state q).
Probabilistic models for each of the states, according to which output strings are
generated upon visiting a state.
Figure 7.9:
GHMM model describing the eukaryotic gene. E states correspond
to exons, while I states correspond to introns.
The probabilistic model for gene structure as suggested by Berge
and Karlin [1], is based on GHMM (see figure 7.9).
The states of the GHMM correspond to the different functional units on a gene,
like promoter regions, exon, intron etc. The transition between
the states ensure that the order in which the model visits various states
is biologically consistent.
The states for an intron and an internal exon are subdivided
according to phase offset to the codon frames. For
,
the state Ii (respectively, Ei) corresponds to
introns (exons) starting i positions after a codon starts. Note
that the only transition from Ii to any internal exon state is
to Ei. Also note that the model is divided into two symmetric
halves. The upper half of the figure (states with a ``+"
superscript) models a gene on the forward strand and the lower
half models a gene on the backward strand of the genomic sequence.
If the parameters (like ,
ai,j, etc.) are suitably
determined, then the model can be used for gene structure
prediction in the following manner.
Definition 7.1
A parse
of sequence S is an ordered sequence of states
(
)
with an associated duration di to each state. The length of
is
Suppose we are given a DNA sequence S and a
parse ,
both of length L. The conditional probability of
the parse
given that the sequence generated is S,
can be computed as:
Let Si be the segment of S produced by qi, and let
P(Si|qi,di)
be the probability of generating Si by the sequence generation model
of state qi with length di.
The most probable parse,
,
can be computed by Viterbi like algorithm. P(S) can be
computed by a forward-like algorithm.
Next:GENSCAN Up:Gene Finding Previous:Gene Structure in EukaryotesItshack Pe`er 1999-02-03