| organism |
test type |
% maps with errors |
| Random |
no postscreening |
|
| Random |
PS 500 probes before |
|
| Random |
PS 500 probes after |
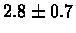 |
| Bacterium |
no postscreening |
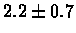 |
| Bacterium |
PS 500 probes before |
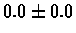 |
| Bacterium |
PS 500 probes after |
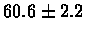 |
| Yeast |
no postscreening |
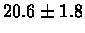 |
| Yeast |
PS 500 probes before |
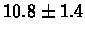 |
| Yeast |
PS 500 probes after |
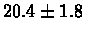 |
| C. Elegans |
no postscreening |
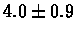 |
| C. Elegans |
PS 500 probes before |
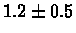 |
| C. Elegans |
PS 500 probes after |
|
| Human |
no postscreening |
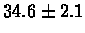 |
| Human |
PS 500 probes before |
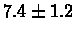 |
| Human |
PS 500 probes after |
|
|