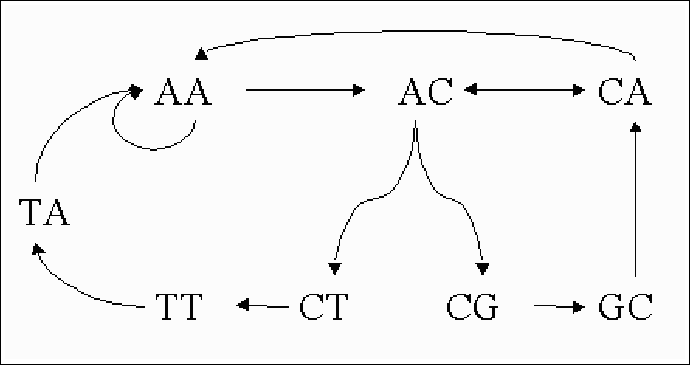
 |
If we take k=8, which is a practical length, and p=0.01, we get that the expected length of the reconstructed sequence is 210 bases. When one considers genes, that can be several thousands of bases long, this is obviously not good enough. Some other chip designs achieve a somewhat better result, but these designs are only theoretical. They are usually very difficult or impossible to manufacture, and cannot be used in true biological context. Sadly, then, sequencing based on hybridization is not a real alternative to standard sequencing.